Late Onset Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes Diagnosed Using Urinary Epithelial Cells
Article information

Abstract
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) together comprise a rare disease of mitochondrial inheritance that usually presents its first symptom before 40 years of age. Here, we report the case of a patient with late onset MELAS, diagnosed using urinary epithelial cells after repetitive negative test results with leukocyte blood samples. A 71-year-old woman visited Dongsan Hospital for aphasia and mild drowsy mentality. Magnetic resonance imaging showed diffusion restriction of the left anterior temporal, parietal, and occipital lobes. We repetitively tested the mitochondrial-encoded gene MT-TL1 deoxyribonucleic acid (DNA) sequencing using blood samples and the results were repeatedly negative. In samples with urine epithelial cells, point mutation m.3243A>G was detected. The level of mutant mitochondrial DNA heteroplasmy in the urine sample was 15%. Our case demonstrates the potency of using urinary epithelial cells in mutation detection for diagnosing late onset MELAS.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes together form a rare disease termed MELAS of mitochondrial inheritance that usually presents its first symptom before 40 years of age.1 The most frequent cause is a mitochondrial deoxyribonucleic acid (mtDNA) mutation (m.3243A>G) in the MT-TL1 gene, which accounts for up to 80% of the MELAS affected population.2,3 Although detection of mtDNA mutation in blood leukocytes is a convenient method for diagnosis, the ratio of mutation is often lower than that in the muscle, which could lead to false-negative results.4 Therefore, when the diagnosis is not confirmed in the blood sample, muscle biopsy is recommended as a routine investigation.5 However, owing to its invasiveness, detecting mutation of mtDNA with urinary epithelial cells could be a better alternative for the diagnosis.2,5 Here, we report a case of late onset MELAS patient diagnosed using urinary epithelial cells after repetitive negative blood test results with leukocytes.
CASE
A 71-year-old woman visited Dongsan Hospital with acute language disturbance and aggravated chronic headache for several days. The patient had a history of diabetes mellitus since 6 years and hypertrophic cardiomyopathy since 1 year. She had also been experiencing headaches and hearing loss for decades. The patient had a petite body type with 150 cm height, 31 kg weight, and body mass index 13.78 kg/m2. She showed diffuse muscular atrophy and had general weakness without lateralized motor symptoms. Neurologic examination revealed mild drowsy mentality with sensory aphasia. Magnetic resonance imaging (MRI) showed diffusion restriction of the left anterior temporal lobe on admission. Follow-up MRI performed during admission showed progression with diffusion high signal involving the left parietal, occipital, and temporal cortex (Fig. 1A). Furthermore, significant vasogenic edema was noticed in this area in the fluid attenuated inversion recovery image (Fig. 1B). MR angiography showed mild stenosis in the left middle cerebral artery M2 segment (Fig. 2). Serum lactic acid level was 1.8 mmol/L, which was within the reference range. An electroencephalogram showed periodic lateralized epileptiform discharge in the left parieto-temporal area. The Korean version of the Western Aphasia Battery revealed severe Wernicke’s aphasia. A transthoracic echocardiogram showed ejection fraction of 57% with hypertrophy of left ventricular walls and thickened papillary muscle. The 24-hour Holter monitoring showed no significant abnormality. Heart dynamic computerized tomography scan revealed no evidence of intracardiac thrombus or shunt. Laboratory test results for thyroid stimulating hormone, antinuclear antibody, anti-neutrophil cytoplasmic antibody panels, anticardiolipin antibody, and complement titers were normal. Cerebrospinal fluid analysis revealed two leukocytes, protein 47.6 mg/dL, and glucose 72 mg/dL. The patient’s daughter died of mitochondrial disease 5 years ago, but the exact name of the disease was not known. No other family members including the patient’s parents, siblings, sons, or close relatives were known to have mitochondrial or genetic diseases.
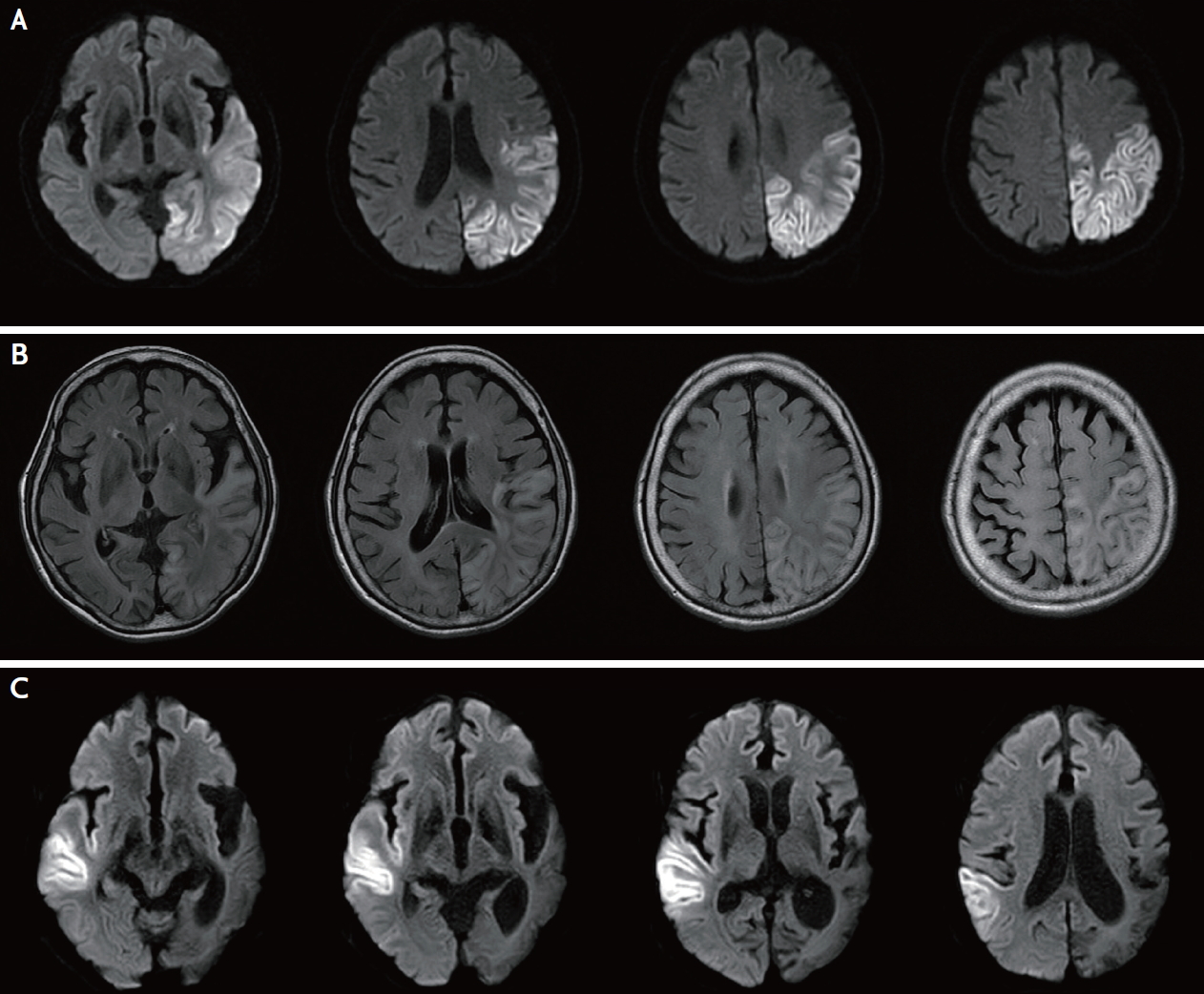
(A) The magnetic resonance imaging of the brain shows diffusion restriction of the left occipital, parietal, and temporal lobes that crosses several vascular territories. (B) Vasogenic edema is observed in the fluid attenuated inversion recovery images. (C) Diffusion restriction of right parietotemporal cortex during second admission.

The magnetic resonance angiography shows mild stenosis of left middle cerebral artery M2 segment (white arrow).
Mitochondrial-encoded gene MT-TL1 DNA sequencing was performed by using blood samples and the test result was negative. Repeated tests still showed negative result. Finally, we decided to retest using urine samples, and point mutation m.3243A>G was detected. The level of mutant mtDNA heteroplasmy in the urine sample was 15%. The patient was treated with single antiplatelet and antiepileptic drugs and transferred to the local hospital when she stabilized. After 2 years, the patient was re-admitted to Dongsan Hospital for deterioration of mental status and clinical seizure. The MRI showed diffusion restriction in the right parietotemporal cortex, which was the opposite side of the previous lesion (Fig. 1C).
DISCUSSION
Generally, the first symptom of MELAS presents before the age of 40 years. Our patient showed onset of clinical symptoms at 71 years, which is one of the oldest ages reported among previous studies. We repeatedly obtained negative results from blood tests, which could be associated with a decrease in the mutation load in blood as the patient aged.6 Additionally, low mutation load appeared to be related to the mild and late manifestation of the patient’s symptoms.7,8 Thus, clinical diagnosis of MELAS should not be overlooked in the context of the patient’s old age.
Moreover, despite histological diagnosis with muscle tissue being the preferred diagnostic method, various types of non-invasive samples can be used to identify mutant mtDNA. Urinary epithelial cells are found in 10–30 mL of the first morning urine with 10 minutes of centifuge and are used in genetic analyses.2,4,9 Several studies have shown that the ratio of mtDNA mutation is significantly higher in urine than in blood.4,8,9 Ma et al.4 reported the mean mutation loads of 36.2% from blood and 63.5% from urine in 11 probands, while Shanske et al.10 found mean mutation loads of 23.5% from blood and 63.0% from urine samples of 32 individuals from families known to harbor the m.3243 A>G mutation. Our case demonstrates the potency of using urinary epithelial cells in the mutation detection for diagnosing late onset MELAS.
This method has certain limitations. There are no standard guidelines for the best urinary sample preparation and this method still does not have very high sensitivity. However, when blood samples of elderly patients suspected of MELAS report repeated negative leukocyte tests, urinary epithelial cells could be useful in diagnosing MELAS.
Notes
Conflicts of Interest
No potential conflicts of interest relevant to this article was reported.